| Accueil | B.Mol | Gen | Web |
MÉTHODES PHYSIQUES DE SÉPARATION ET D'ANALYSE ET MÉTHODES DE DOSAGE DES BIOMOLÉCULES

6-LA CHROMATOGRAPHIE LIQUIDE / MÉCANISMES ET MODALITÉS
6-2-Les différents modes de chromatographie liquide
On dispose d'un grand nombre de modes de chromatographie.
Certains ont un champ d'application très large, d'autres au contraire
correspondent à des applications très spécialisées.
On distinguera dans ce qui suit :
1 - chromatographie d'adsorption
2 - chromatographie de partage (en phase directe (2A) ou inverse (2B))
3 - chromatographie d'échange d'ions (+ chromatographie ionique)
4 - chromatographie de paires d'ions
5 - chromatographie d'exclusion (séparation selon la taille)
6 - chromatographie d'affinité
7 - chromatographie adaptée à la séparation d'énantiomères
Le choix de tel ou tel mode obéit à des règles logiques directement liées à la nature des composés à séparer :

6-2-1-La chromatographie d'adsorption
C'est la première chromatographie réalisée : séparation des pigments végétaux par adsorption sur de la craie (Tswett 1906).
Définition : Ce mode de chromatographie met en jeu un mécanisme d'adsorption du soluté sur la phase stationnaire solide et un mécanisme d'élution (désorption) par la phase mobile liquide ou gazeuse (éluant).
Principe :
- L'adsorption est la fixation plus ou moins énergique d'un gaz, d'un liquide ou d'un soluté sur une surface solide; elle met en jeu des liaisons à faible énergie (liaisons hydrogène, interactions électrostatiques...). Pour être utilisable à des fins séparatives, l'adsorption doit être réversible.
- L'élution ou désorption consiste à extraire le soluté adsorbé à l'aide d'un solvant appelé éluant.
- Les différents solutés sont plus
ou moins adsorbés sur la phase stationnaire, et plus ou moins solubles
dans la phase mobile; il en résulte une migration différentielle
des solutés en fonction de la résultante entre les deux
forces (de rétention et d'entraînement) et donc une séparation
de ces solutés.
Le schéma ci-après résume les interactions entre
le soluté, le solide adsorbant et l'éluant :

Adsorbants :
- Les adsorbants doivent être insolubles
dans le solvant et chimiquement inertes
vis-à-vis du solvant et des solutés.
- Leur granulométrie varie
entre 5 et 100 µm et leur surface spécifique
de 50 à 1000 m2/g,
- L'activité d'un adsorbant dépend de sa nature et de sa
teneur en eau.
N. B. Dans certains cas, on cherche
à diminuer l'activité adsorbante, et on fait au contraire
une désactivation.
-La capacité d'adsorption
peut être :
- faible (carbonate de calcium...)
- forte (silice, alumine, charbon ...)
-La polarité
peut être
- faible (charbon...)
- forte (silice SiO2, utilisée sous forme hydratée : gel de silice, qui présente des fonctions "silanol" : Si-OH , alumine Al2O3, n H2O...)
Solvants :
On utilise généralement des mélanges de solvants,
de polarité voisine de celle des solutés à séparer
(ceux-ci doivent être solubles dans l'éluant !). On classe
les différents solvants selon leur "force éluante" qui traduit
leur polarité:
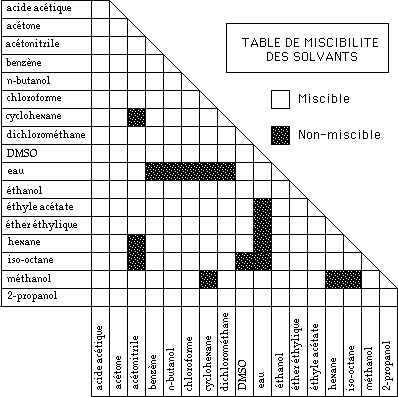
Série éluotrope sur alumine de quelques solvants, classés par ordre de polarité croissante :
|
Selon la nature des composés à séparer,
on choisit des solvants de force éluante (![]() o)
appropriée. C'est rarement un solvant pur, mais en fait un mélange
de 2 à 4 solvants, optimisé par tests successifs vis-à-vis
des composés étudiés, sans que cela obéisse
à des règles permettant de prédire les résultats.
Des abaques permettent de calculer la force éluante d'un mélange
binaire :
o)
appropriée. C'est rarement un solvant pur, mais en fait un mélange
de 2 à 4 solvants, optimisé par tests successifs vis-à-vis
des composés étudiés, sans que cela obéisse
à des règles permettant de prédire les résultats.
Des abaques permettent de calculer la force éluante d'un mélange
binaire :

Dans les mélanges contenant plus de deux solvants, les derniers sont présents dans de très faibles proportions (< 2%) et correspondent à des solvants de forte polarité (eau, acide) destinés à améliorer la symétrie des pics. Le choix du solvant doit obéir à certaines contraintes :
- miscibilité
des composantes élémentaires |
* N.B. Cette contrainte n'existe pas avec la chromatographie sur couche mince, pour laquelle le choix des solvants est donc plus étendu.
Applications :
La chromatographie d'adsorption est utilisée pour séparer des molécules organiques de masse molaire 100 à 1000 g/mol et de polarité moyenne;
-les molécules apolaires sont pas ou peu retenues, et sont éluées les premières (= elles migrent au front en CCM).
-Les molécules trop polaires risquent d'être adsorbées de façon irréversible, et ne sont donc pas éluées (= elles resteraient sur la ligne de dépôt en CCM).
Exemples :
- lipides : stérols et stéroïdes, caroténoïdes, phospholipides...
- pigments, médicaments...
- oses et oligosides, acides aminés et oligopeptides
L'utilisation de ce mode de chromatographie est assez large, en raison de sa facilité d'emploi et du fait que la transposition depuis la chromatographie sur couche mince est très aisée.
 |
6-2-2-La chromatographie de partage
La chromatographie de partage (ou chromatographie liquide-liquide) met en jeu un mécanisme de partition entre solvants que constituent respectivement par la phase stationnaire et la phase mobile. La phase stationnaire peut être constituée par un film liquide (non miscible avec la phase mobile bien sûr) imprégné sur un support rigide (silice) ou fixé par liaison covalente (phases greffées). C'est ce second type qui est utilisé actuellement. Le greffage est réalisé par établissement de ponts siloxane (Si-O-Si) :
->Si-OH + X-Si(CH3)2-R --> ->Si-O-Si(CH3)2-R + HX
Selon la nature des radicaux R, on distinguera :
- les phases greffées polaires (-diol, -cyanopropyl, aminopropyl,…)
- les phases greffées apolaires (-alkyl, -phényl, …)
Les premières sont utilisées avec des
solvants peu polaires (cf adsorption) et permettent de réaliser
de la chromatograhie en phase
normale ou directe.
Les secondes s'emploient avec des solvants polaires et l'on a alors de
la chromatographie en phase inverse ou réverse.
En première approximation, l'ordre d'élution des composés
est inversé entre les deux modes, les composés polaires
étant élués en premier dans les systèmes en
phase réverse.
LES PHASES GREFFEES
POLAIRES
Elles s'utilisent comme les colonnes de silice précédentes.
La présence de fonctions particulières sur le greffage (-CN,
-NH2,…) peut apporter une sélectivité intéressante.
De plus, il est possible de travailler avec un gradient
de force éluante, donc d'analyser en une seule fois des mélanges
contenant des composés de polarités très différentes.
Il n'y a pas de phénomènes d'adsorption irréversible
et on peut rincer les colonnes avec des solvants très éluants
comme du méthanol pur.
LES PHASES GREFFEES APOLAIRES
Ce sont les supports les plus couramment utilisés, principalement
avec un greffage alkyl (octadécylsilane ou -ODS ou -C18). Dans
ces systèmes, le solvant le plus polaire (l'eau) est le moins éluant
et la force éluante de la phase mobile est augmentée par
ajout d'un solvant organique (méthanol, acétonitrile).
La phase stationnaire (type de greffage, taux de greffage, porosité)
et la phase mobile (type de solvant organique, pH, température,…)
concourent toutes deux à la sélectivité de
l'ensemble et les possibilités sont innombrables, ce qui explique
la popularité de ce mode chromatographique.
Le contrôle de pH permet de faire varier à volonté
le k' des composés ionisables. Il convient dans le cas de greffage
sur silice de rester dans des limites compatibles avec la non-dissolution
de la colonne ( 2 < pH < 8 ). Il existe par ailleurs des phases
greffées sur des résines polymérisées qui
n'ont pas cet inconvénient et permettent de travailler dans une
gamme de pH bien plus large (1-13).
Certains greffages comme le greffage -CN ont des propriétés
hybrides qui permettent de les utiliser aussi bien en phase directe qu'en
phase inverse.
![]()
6-2-3-La chromatographie d'échange d'ions
Définitions : Dans la chromatographie d'échange d'ions, la phase stationnaire comporte des groupements ionisés (+ ou -) fixes; des ions mobiles de charge opposée assurent l'électroneutralité. Les ions retenus au voisinage des charges fixes sont échangeables avec les ions présents dans la phase mobile.
La séparation est basée sur cette propriété d'échange d'ions et ne peut donc s'appliquer qu'à des solutés ionisables.
Les échangeurs d'ions :
Nature :
La phase stationnaire est constituée d'un support insoluble dans l'eau, sur lequel sont greffés des groupements fonctionnels ionisables.

-Les supports peuvent être :
• minéraux : silice
• organiques : résine polystyrénique, cellulose, dextrane
-Les groupements fonctionnels
sont fixés par des liaisons covalentes sur la matrice; ils sont
de deux types :
• les échangeurs de cations portent des groupements chargés (-)
• les échangeurs d'anions portent des groupements chargés (+)
Exemples :
| ECHANGEURS DE CATIONS |
ECHANGEURS D'ANIONS |
|||
| FORTS
FAIBLES |
||||
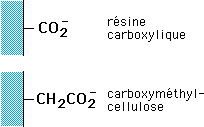 |
 |
|||
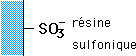
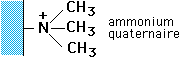
Les échangeurs forts sont ionisés quel
que soit le pH, alors que les carboxylates ne le sont qu'à pH >
3 et les amines tertiaires ne sont protonées qu'à pH acide.
On utilise les échangeurs d'anions forts pour séparer des
acides faibles (ex acides carboxyliques) et les échangeurs faibles
pour séparer des acides forts (ex esters phosphate). Le même
type de raisonnement s'applique aux échangeurs de cations.
La nature de l'ion échangeable est variable : par exemple une résine
cationique peut être utilisée sous forme acide (liée
à H+) ou sous forme sodique (liée à Na+);
il faut donc conditionner la
résine sous la forme ionique adéquate avant son utilisation,
et la régénérer
après usage.
Caractéristiques
:
-Porosité : le support
est un polymère plus ou moins réticulé ; la porosité
dépend du taux de pontage : un polymère très réticulé
a des pores de petite taille et convient pour des petites molécules.
-Granulométrie : le support
est commercialisé sous forme de grains de diamètre variant
de 30 à 800 µm (chromatographie basse pression) Celui des colonnes
HPLC a une granulométrie de 5-10 µm (supports totalement poreux)
ou légèrement supérieure (supports pelliculaires,
en couche de 1 µm sur des billes de verre). Les échanges sont d'autant
plus rapides et efficaces que les grains sont petits.
-Capacité de rétention
: c'est la quantité maximale d'ions que peut fixer l'échangeur
d'ions, exprimée en mEq/g de poids sec ou en mEq/ml de poids humide.
Elle dépend de la densité du support en groupements fonctionnels
et il faut en tenir compte pour ne pas trop charger une colonne.
L'affinité d'un échangeur pour un ion donné dépend de plusieurs facteurs:
• de la charge des groupements fonctionnels, qui elle-même dépend :
L'affinité d'un échangeur
pour un ion donné dépend de plusieurs facteurs:
• de la charge des groupements fonctionnels, qui elle-même dépend :
- de leur nature (pKa)
- du pH de la solution
• de la densité de charge de l'ion considéré, qui dépend:
- de la charge de cet ion c'est-à-dire :
- de la nature de l'ion (pKa, pHi)
- du pH de la solution
- de la taille de l'ion (un ion est d'autant plus fixé qu'il est chargé, ou/et qu'il est petit)
• de la concentration des ions
• de l'accessibilité des groupements
fonctionnels ( cf : porosité et granulométrie)
Réaction d'échange d'ions :

Le système peut s'écrire : ![]() (r = fixé sur la résine; s = en solution)
(r = fixé sur la résine; s = en solution)
La constante de sélectivité est donnée par la relation :
![]()
et
si ![]() > 1 la résine a une affinité préférentielle
pour le soluté A
> 1 la résine a une affinité préférentielle
pour le soluté A
si ![]() < 1 la résine a une affinité préférentielle
pour le soluté B
< 1 la résine a une affinité préférentielle
pour le soluté B
Les éluants
sont des solutions aqueuses qui contiennent des ions échangeables
avec les solutés fixés sur l'échangeur :
-solutions contenant un ion de densité de charge plus élevée
et (ou) de concentration plus élevée.
-solutions d'un pH tel qu'il modifie la charge des ions fixés et
(ou) des groupements fonctionnels et provoque leur libération dans
l'éluat.
On peut éluer avec une solution de composition constante (conditions
isocratiques) ou avec un gradient de pH et (ou) de force ionique, pour
décrocher successivement les différents ions fixés
sur l'échangeur.
Applications : La chromatographie d'échange d'ions est utilisée pour séparer des molécules ionisables, quelle que soit leur taille : ions minéraux, acides aminés, peptides, protéines, nucléotides, acides nucléiques, glucides ionisés et lipides ionisés.
![]()
6-2-4-La chromatographie de paires d'ions
On appelle paire
d'ions une entité formée par l'association de
deux ions de charge opposée. La paire d'ions a donc une charge
nette nulle et peut dans ce cas présenter des propriétés
± hydrophobes qui permettent de l'analyser par sur une colonne de chromatographie
en phase inverse.
Pour arriver à cette utilisation, on emploie des phases mobiles
qui contiennent un détergent anionique
ou cationique. Ces détergents sont des molécules
ionisées qui comportent par ailleurs des chaînes carbonées
hydrophobes (ex tétrabutylammonium, laurylsulfonate,…) et
forment avec les composés à séparer des paires d'ions
hydrophobes.
On peut dans ce mode chromatographique jouer sur un très grand
nombre de paramètres :
- nature et concentration du détergent
- pH et force ionique du tampon
- température
- modifiant organique (alcool, acétonitrile,…)
De tels systèmes s'avèrent plus performants (N) que les systèmes d'échange d'ions et ont des capacités plus grandes ( = ils permettent d'injecter de plus grandes quantités d'échantillon sur une colonne de même dimension).
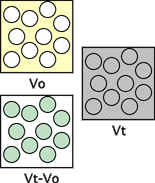
6-2-5-La chromatographie d'exclusion
La chromatographie d'exclusion est aussi
appelée FILTRATION SUR GEL
ou TAMISAGE MOLECULAIRE. Cette
technique est apparue en 1959 avec un produit nouveau : le Sephadex.
Le Sephadex est un gel de dextrane
(polymère de glucose a-1-6 produit par Leuconostoc mesenteroïdes),
auquel on fait subir une réticulation. Il se présente sous
forme de billes poreuses, dont la porosité dépend du degré
de réticulation. Ces billes sont très hydrophiles et gonflent
dans l'eau. Il en existe différents types en fonction de la taille
des billes et de leur porosité.
Principe de la chromatographie d'exclusion :
|
|||||||
| 1
: Dépôt d'un mélange de deux molécules
(des grosses et des petites) sur une colonne remplie d'un gel
de Sephadex. |
|||||||

D'une façon plus générale :
-Les molécules
de taille supérieure à celle des pores des billes
de Sephadex sont totalement exclues du gel et ne se répartissent
que dans le volume extérieur aux billes, c'est-à-dire
dans la phase mobile (éluant). Elles sortent les premières,
à un volume d'élution Vo appelé volume mort de
la colonne.
-Les molécules de taille inférieure
à celle des pores des billes de Sephadex peuvent pénétrer
librement dans les billes et se répartissent dans l'ensemble
des liquides de la colonne (volume de liquide à l'intérieur
des billes ou phase stationnaire + volume de liquide à l'extérieur
des billes ou phase mobile). Elles sortent donc les dernières,
à un volume d'élution Vt ou volume total des liquides
de la colonne.
-Les molécules de taille intermédiaire
pénètrent un peu dans les billes, en fonction de leur
taille et de leur forme; elles pénètrent d'autant moins
qu'elles sont plus grosses, et elles sont éluées dans
l'ordre des masses molaires décroissantes.

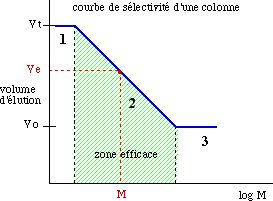
-Cette technique est également appliquée au FRACTIONNEMENT de MELANGES de MACROMOLECULES et à la DETERMINATION (approximative) de la MASSE MOLAIRE des protéines. Dans ce dernier cas, il faut d'abord étalonner la colonne avec des protéines de masse molaire connue, tracer la courbe Ve = f(logM), puis effectuer une détermination graphique.
Ce qui précède concernant le Sephadex est en fait très général et s'applique à de nombreux supports, en particulier des supports à base de silice ou de polymères organiques rigides, qui permettent l'utilisation de ces supports en HPLC (ce n'est pas le cas du Sephadex). Ces colonnes, le plus souvent de taille réduite par rapport aux précédentes, mais également plus résolutives et donnant des séparations plus rapides (car autorisant l'emploi de débits linéaires beaucoup plus élevés) sont utilisées à des fins analytiques :
- avec des phase mobiles aqueuses pour les
macromolécules biologiques (essentiellement protéines)
et avec des phases stationnaires conçues pour minimiser les
phénomènes d'adsorption et de dénaturation.
- avec des phases mobiles organiques pour l'analyse des polymères
en chimie organique (ex polystyrènes).
6-2-6-La chromatographie d'affinité
Ce mode de chromatographie connaît depuis 1970 un développement sans précédent et est appelé à prendre une place encore plus grande avec l'essor des biotechnologies.
Le principe consiste à utiliser une phase stationnaire constituée d'un support (silice, polymère) sur lequel on a greffé une molécule organique particulière qui présente une affinité sélective pour certains constituants d'un mélange dont on cherche à les isoler. Ceux-ci vont être sélectivement adsorbés ou tout au moins retenus sur la colonne, tandis que les autres composants sont très rapidement élués. Un changement de la phase mobile (pH, force ionique ou ajout d'un compétiteur) permet ensuite d'éluer les substances intéressantes, avec un facteur de purification pouvant atteindre 1000.
La chromatographie d'affinité s'utilise avec des colonnes basse pression ou des supports à haute performance (HPLAC). Nous allons illustrer cette technique avec quelques exemples très classiques ou moins courants.
Techniques en basse pression :
- isolement des ARNm polyA+ sur colonne
d'oligodT- ou polyU- sepharose à partir d'un extrait d'ARN
total (les ARNm représentent 1% du total)
- purification de récepteurs hormonaux avec des colonnes sur
lesquelles on a fixé des molécules d'hormones
- à un degré de spécificité très
inférieur, les colonnes sur lesquelles sont greffées
des ions boronate ont une affinité pour les diols cis et permettent
de fixer entre autres les sucres.
Techniques en haute pression (à titre d'exemple) On donnera ici comme exemple le choix offert par un fabricant (Pierce Eurochemie) :
| Type de phase stationnaire |
Type d'application |
| Protéine A (de Staphylococcus aureus) |
Immunoglobulines |
| Bleu Cibacron |
Enzymes (ex. déshydrogénases) |
| Boronates (affinité pour diols 1,2 ou 1,3) |
Nucléosides, nucléotides, ARNt, Glycoprotéines, sucres |
| Concanavaline A (lectine) |
Glycoprotéines, sucres |
| Trésyle activé (pour faire sa colonne) |
Sert à fixer amines primaires et thiols |
6-2-7-Techniques pour la séparation d'énantiomères
La séparation d'isomères
optiques est un problème qui se pose souvent en biologie, où
beaucoup de molécules possèdent un (ou plusieurs) carbone(s)
asymétrique(s) : exemple des acides aminés, des hydrocarbures
insaturés,...
La synthèse chimique, pour sa part, conduit souvent à
des mélanges d'isomères qu'il faut ensuite de séparer
afin d'isoler le composé biologiquement actif (exemple des
phéromones). Or ces substances ont des propriétés
chimiques souvent très voisines et ne sont que mal séparées
par les techniques de chromatographie liquide conventionnelles. On
a recours alors à des méthodes plus sophistiquées
qui font souvent appel à l'utilisation de substances optiquement
actives, soit dans la constitution de la phase stationnaire, soit
mises en solution dans la phase mobile. Nous nous bornerons ici à
donner quelques exemples, les possibilités étant en
fait très nombreuses.
![]()
Séparation des aminoacides D et L
Le mode de chromatographie utilisé est celui dit de "l'échange de ligandes". On met à profit l'existence d'une formation de complexes entre les métaux de transition (Cu, Zn, Cd, Ni) et les aminoacides.
- dans le mode statique, la phase stationnaire est greffée avec de la L-Proline par l'intermédiaire d'un bras et forme un complexe avec des ions Cu++. Cette structure qui comporte deux carbones asymétriques est hautement stéréosélective et interagit de façon très différente avec les aminoacides des séries D et L, qui sont donc retenus de façon très distincte.
- dans le mode dynamique, la phase stationnaire est un échangeur de cations et contient des ions Cu++ ainsi que de la L- ou D-proline. Les complexes formés sont retenus différemment des formes non complexées, et ceci a pour conséquence de séparer les formes D et L des aminoacides injectés sur la colonne.
Séparation d'isomères en utilisant des complexes d'inclusion
Cette méthode utilise des cyclodextrines (polyosides cycliques formés de 6, 7 ou 8 unités glucose) formant une structure torique au sein de laquelle certaines molécules peuvent s'inclure de façon réversible et d'une façon qui dépend fortement de leur structure spatiale.
- dans le mode statique, la méthode utilise des phases greffées avec des cyclodextrines
- dans le mode dynamique, on dissout une cyclodextrine dans le solvant.
Dans un certain nombre de cas, cette méthode
s'avère très efficace pour séparer des isomères.
Ce mode de chromatographie est encore peu utilisé, mais il
est en fait assez récent (surtout l'usage des cyclodextrines)
et son utilisation devrait se développer assez rapidement.
